
La troisième génération de séquenceurs
Dans une news de Nature Check Hayden, Erika. (2008) rapporte qu'à une conférence Advances in Genome Biology and Technology en Floride le 5 février, on a pu voir la troisième génération de séquenceurs et qu'ils promettent le génome humain à $5,000 encore en 2009.La 3ème génération ? j'ai dû manquer la 2ème ?
La 2ème appelée aussi next-generation a été décrite dans un bio-tremplin précédent et a été utilisé pour séquencer un génome humain entier, diploïde. Notamment la technique du pyrosequençage 454.Certains parient sur la vitesse.
A cette conférence, Complete Genomics ont montré un génome qu'ils ont séquencé en 8 jours avec 9 machines : Ils annoncent avoir séquencé 254 gigabases (254 milliards de paires de bases) et couvert 92% du génome d'un homme anonyme. En effet les séquenceurs les plus rapides séquencent des fragments très courts (quelques dizaines) par rapport à la classique méthode Sanger et ses dérivés (plusieurs centaines). Il faut donc séquencer plusieurs fois chaque base : on parle de profondeur de séquençage : ils ont lu chaque base 91 fois. Selon la compagnie, ils ont un taux d'erreur de mon d'un tiers de pourcent.Ils commenceront à vendre leurs services et promettent le génome humain à 5'000$ cette année encore.
Certaines régions sont très difficiles à séquencer car elles sont répétitives ce qui explique qu'on n'ait pas la totalité du génome. C'est aussi une difficulté avec les autres techniques et on considère souvent que ces séquences sont peu importantes et le projet génome humain a té déclaré achevé alors qu'on n'avait pas encore absolument tout séquencé.
D'autres parient plutôt sur la longueur et la précision.
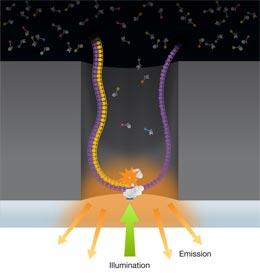
Figure 2 : Source : Check Hayden, Erika. (2008) Nature news
D'autres compagnies parient sur la longueur du fragment séquencé comme Pacific Biosciences avec une technique qui lit au fur et à mesure que la polymérase avance sur une seule molécule d'ADN - mais actuellement à la vitesse de 3 bases par seconde (les autres atteignent 30'000 bases / seconde) - et sur des longueurs beaucoup plus grandes (en moyenne 586 paires de bases, et jusqu'à 2,805). Ils déclarent une précision de 99.9999% avec une profondeur de lecture de 38 sur un génome complet de Escherichia coli. Ils promettent un génome humain en 3 minutes pour 2013.

Figure 3 : Chaque fois que la polymérase fixe une base, un flash de lumière colorée est émis depuis un puits minuscule et détecté. Une caméra permet de suivre la séquence en cours de formation. Figure complète ici Source : Eid, John., et al. (2009)
Mais où s'arrêteront-ils ?
Ils ne s'arrêteront sans doute pas. Et les conséquences de la disponibilité aisée et bon marché de séquençage en masse sont déjà en train de changer la recherche, bien sûr, mais aussi la médecine, et a des conséquences en droit et en économie. Nous en verrons dans les bio-tremplins. Voyons un exemple très récent en écologie.L'écologie par les technologies de l'information ?
 On a découvert récemment rapporte Zelkowitz, Rachel.(2009) qu'une guêpe braconide parasite qui pond ses oeufs dans des chenilles utilise pour paralyser ses proies une toxine qui vient d'un virus (Polydnaviruse) qui a infecté ces guêpes il y a quelques millions d'années et y est resté. L'ADN du virus est intégré au génôme de la guêpe et produit des virus qui sont injectés avec les oeufs de la guêpe dans la chenille et neutralisent ses défenses immunitaires.
On a découvert récemment rapporte Zelkowitz, Rachel.(2009) qu'une guêpe braconide parasite qui pond ses oeufs dans des chenilles utilise pour paralyser ses proies une toxine qui vient d'un virus (Polydnaviruse) qui a infecté ces guêpes il y a quelques millions d'années et y est resté. L'ADN du virus est intégré au génôme de la guêpe et produit des virus qui sont injectés avec les oeufs de la guêpe dans la chenille et neutralisent ses défenses immunitaires.Les OGM des millions d'années avant l'heure en somme.
Le virus est incapable de se multiplier autrement que dans les ovaires de la guêpe, et la guêpe a besoin du virus pour se reproduire. Les auteurs parlent d'une forme de symbiose obligatoire entre le virus et la guêpe.
Une nouvelle biologie vient élargir notre discipline
Ce qui mérite d'être souligné ici c'est que l'étude écologique de cette espèce passe par une exploration du génome : c'est en séquençant le génome du virus et une partie de celui de la guêpe, puis en les comparant avec des outils bioinformatiques de recherche de séquences similaires ( Comme BLAST) d'alignement et de comparaison de séquence désormais omniprésents qu'on a élucidé le mystère (Bézier, Annie, et al. 2009) Ces outils sont désormais largement utilisés dans tous les domaines de la biologie, même ceux où on ne les attendrait pas, la biologie de terrain, la botanique ou l'écologie. Ils y passent 50-70% de leur temps selon mes sondages...On assiste donc au développement d'un nouvelle forme de biologie parfois appelée in silico qui complète et élargit la biologie in vitro et in vivo. Et les technologies de traitement de l'information (BIST) prennent une place grandissante dans l'activité d'un biologiste à travers tous les domaines. Faut-il intégrer dans ces chapitres de la biologie les nouvelles façons de les pratiquer, faut-il y former les élèves ?
Sources
- Bézier,Annie et al. (2009). Polydnaviruses of Braconid Wasps Derive from an Ancestral Nudivirus. Science 13 February 2009: Vol. 323. no. 5916, pp. 926 - 930 DOI: 10.1126/science.1166788
- Check Hayden, Erika. (2009). Genome sequencing: the third generation Nature News 6 February 2009 | doi:10.1038/news.2009.86
- Check Hayden, Erika. (2008). Human genomes in minutes? Not yet, but biotechnology company is on track for 2013. Nature news 20 November 2008 | doi:10.1038/news.2008.1245
- Eid, John., et al. (2009). Real-Time DNA Sequencing from Single Polymerase Molecules, Science 323 (5910), 133. DOI: 10.1126/science.1162986]
- Espagne, Eric et al. (2004) Genome Sequence of a Polydnavirus: Insights into Symbiotic Virus Evolution Science 306 (8 October 2004) (5694), 286. [DOI: 10.1126/science.1103066]
- Zelkowitz,Rachel. (2009) Ancient Virus Gave Wasps Their Sting ScienceNOW 12 February 2009





Pour la 3ème génération, ça dit que "ils ont un taux d'erreur de moins d'un tiers de pourcent. ". ça sonne comme un exploit, mais sur 254 gigabases, ça fait toujours plus de 750 millions d'erreurs!! On peut se permettre un telle imprécision? Quel est le critère d'évaluation pour ça?
RépondreSupprimerM.T. Via F.lo
Patrick Descombes, responsable de la plateforme Génomique de l'uniGe :
RépondreSupprimerces instruments ont en effet un taux d'erreur qui peut paraitre élevé. Mais il ne faut pas oublier que l'on séquence de manière aléatoire des fragments d'environ 50 à 100 bases, jusqu'à 400 pour 454. Donc on a un immense chevauchement des fragments, ce qui permet d'éliminer les erreurs. Il est clair cependant que des améliorations dans la précisions seront bienvenue et sont en cours, les compagnies travaillant sur des améliorations de la chimie impliquée entre autre.
Christophe HUBERT Responsable Plateforme Génome Transcriptome Bordeaux.
RépondreSupprimerEn effet un taux de 1/3de pourcent (0.33%) semble énorme. Cependant il faut normaliser ce taux d'erreur par rapport à la profondeur de lecteur (nb de lecture pour une bases) soit 91x.